CISTINOSIS Y SÍNDROME DE FANCONI. REPORTE DE CASO. FUNDACIÓN CLÍNICA INFANTIL CLUB NOEL
Angie Milena Cárdenas Silva1, Martha Isabel Carrascal2, Alfonso María Valencia3, Nedier Pedraza4, Gastón Edgardo Castillo5
Resumen
El Síndrome de Fanconi (SF) primario es una entidad poco frecuente, su principal causa es la cistinosis, ocasionada por la mutación en el gen CTNS el cual es responsable de la síntesis de la cistinosina, transportador lisosomal de cistina, lo que permite la acumulación tisular de la misma con manifestaciones clínicas renales y extrarrenales. Se presenta un caso de cistinosis nefropática infantil asociada a SF cuya confirmación diagnóstica se realizó mediante la medición de cistina en leucocitos.
Palabras clave: cistinosis; lactante; Síndrome de Fanconi
CYSTINOSIS AND FANCONI SYNDROME. CASE REPORT. FUNDACION CLINICA INFANTIL CLUB NOEL.
Abstract
The primary Fanconi Syndrome (FS) is a rare entity, its main cause is cystinosis, caused by the mutation in the gene CTNS which is responsible for the synthesis of cystinosine, cystine lysosomal transporter, which allows tissue accumulation of cystina in renal and extrarenal tissue clinical manifestations. We present a case of childhood nephropathic cystinosis associated with FS whose diagnostic confi rmation was made by measuring cystine in leukocytes.
.
Keywords: cistinosis; infant; Fanconi Syndrome
_______________________
1 Médico residente, Especialización en Pediatría Universidad Libre. Miembro GRINPED. Cali, Colombia.
2 Médico pediatra, Especialista en Nefrología. Fundación Clínica Infantil Club Noel. Cali, Colombia.
3 Médico, Especialista en Epidemiología clínica. Docente Especialización en Pediatría. Universidad Libre. Miembro GRINPED. Cali, Colombia.
4 Médico pediatra, Especialista en Nefrología. Fundación Clínica Infantil Club Noel. Cali, Colombia.
5 Médico pediatra, Especialista en Cuidado Intensivo. Fundación Clínica Infantil Club Noel. Docente Especialización en Pediatría. Universidad Libre. Miembro GRINPED. Cali, Colombia.
________________________
Introducción
El SF es una tubulopatía proximal caracterizada por la presencia de poliuria, fosfaturia, glucosuria, proteinuria, acidosis metabólica, retardo de crecimiento y raquitismo. Su causa principal es la cistinosis, la cual representa hasta el 20% de los casos de trastornos tubulares hereditarios (1) con prevalencia estimada es 1: 200.000 nacidos vivos (2).
La cistinosis es una alteración del almacenamiento lisosomal, tiene herencia autosómica recesiva, ocurre por la mutación en gen CTNS (17p13.2) codificante de la cistinosina, condición que lleva a la acumulación intracelular de cristales de cistina. Este depósito produce disfunción enzimática con afectación renal y extrarrenal y enfermedad multiorgánica progresiva (3). Tiene tres formas de presentación: nefropática infantil, nefropática juvenil y la forma ocular no nefropática (4). La cistinosis nefropática infantil es la más frecuente y severa, representa el 95% de todos los casos y tiene sus manifestaciones clínicas durante el primer año de vida (5).
Se detalla el caso de una lactante atendida en la Fundación Clínica Infantil Club Noel en la ciudad de Cali (Colombia), que debuta con la forma nefropática infantil de la enfermedad, para lo cual se cuenta con la aprobación del comité de ética institucional y consentimiento informado de los padres.
Presentación del caso
Femenina afrodescendiente de 11 meses de edad, consulta por cuadro clínico de tres meses de fiebre subjetiva, náuseas, falla en el medro e hiporexia. Cuatro días previos al ingreso presenta deposiciones diarreicas no disentéricas.
Antecedentes. Perinatales: producto de la primera gestación, embarazo de 37 semanas, parto eutócico, peso al nacer 3100 gramos, talla 51 cm. Patológicos: bronquiolitis a los tres meses de edad. Familiares: asma en
tío paterno. Padres no consanguíneos. Revisión por sistemas: poliuria, polidipsia.
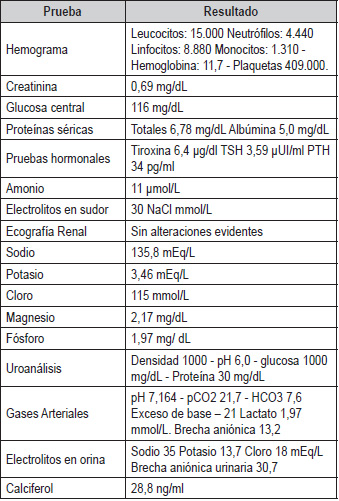
Al examen físico, frecuencia cardiaca 118 latidos por minuto; frecuencia respiratoria 25 respiraciones por minuto; temperatura 36,5 º; SaO2 95% FiO2 21%; peso 6,9 kg; talla 71 cm. Antropometría: peso para la edad -3 desviaciones estándar (DE); talla para la edad 0 y -1 DE; peso para la talla entre -2 y -3 DE; desarrollo neurológico acorde para la edad. Se diagnostica síndrome febril persistente, desnutrición crónica, enfermedad diarreica aguda. Se inicia tratamiento y se solicitan paraclínicos (Tabla 1), los cuales apoyan el diagnóstico de tubulopatía proximal tipo SF.
Se realiza reposición de bicarbonato y fósforo; por persistencia de acidosis metabólica y trastorno hidroelectrolítico, requiere manejo en cuidado intermedio; continúa reposición de electrolitos, fosfato de potasio, bicarbonato y solución de Shohl.
Evolución clínica favorable: de manera progresiva mejora trastornos electrolíticos; la acidosis metabólica persiste; cifras de presión arterial sobre el percentil 95; para la edad se maneja con calcio antagonista y diurético tipo tiazida. Egresa de la institución con prescripción de hidroclorotiazida, amlodipino, ácido fólico, sulfato ferroso, calcio más vitamina D, sulfato de zinc, solución de fosfato, solución de Shohl, bicarbonato de sodio e ion K. Se realiza identificación de cistina en leucocitos por espectrometrías de masas en tándem (1,4 nmol ½ cis/mg proteína) prueba confirmatoria del diagnóstico de cistinosis y se inicia tratamiento con bitartrato de cisteamina.
El estudio genético se realizó mediante Next Generation Sequencing (NSG), el ADN fue extraído a partir de sangre mediante MagNAPure (Roche), ampliación mediante dos primeros específicos para la detección de la deleción de 65 kb. La secuenciación en doble sentido de los productos de amplificación obtenidos mediante secuenciador automático ABI3130XL de Applied Biosystems, estudio posterior de la secuencia mediante software SeqScape v.2.5 y búsqueda de las posibles variantes en la secuencia en distintas bases de datos. Identificando una variante missense patogénica en CTNS: c.926G>T p.G309V (NM_004937) en estado de homocigosis.
Fuente: elaboración propia |
Tabla 1. Exámenes de laboratorio. |
Discusión
Presentamos el caso de una lactante que debuta con manifestaciones clínicas inespecíficas como hiporexia,
náuseas, falla en el medro, polidipsia, poliuria, alzas térmicas subjetivas. Al examen físico la antropometría es compatible con desnutrición, los estudios paraclínicos evidencian acidosis metabólica con brecha aniónica normal, trastornos electrolíticos, uroanálisis con hipostenuria y glucosuria, glucosa central normal. Estos hallazgos sugieren tubulopatía proximal, confirmando la cistinosis con la medición de cistina en leucocitos.
La importancia del enfoque y el diagnóstico oportuno en una condición clínica con manifestaciones sutiles es el tratamiento temprano y el impacto en la calidad de vida del paciente. La literatura disponible acerca de esta entidad clínica es limitada y por lo tanto la caracterización de un caso brinda herramientas a la comunidad académica y médica en su actuar diario.
La cistinosis nefropática juvenil es la causa más frecuente de SF primario, la mayoría de los pacientes con esta patología son asintomáticos al nacimiento y su desarrollo es normal hasta los 6 meses de edad (6); la historia natural de la enfermedad se orienta hacia la insuficiencia renal terminal en la primera década de la vida, siendo la diálisis y el trasplante alternativas disponibles. El caso descrito evidencia el compromiso nutricional, polidipsia, poliuria como primera manifestación y, posteriormente, los trastornos electrolíticos que llevan a la descompensación clínica.
CTNS es el gen que codifica la cistinosina, una proteína lisosomal transmembrana que actúa como simportador protón cistina, aprovechando el gradiente de protones para transportar este aminoácido desde el lumen lisosomal hasta el citosol. La disfunción de este sistema lleva a acumulación de la cistina en forma de cristales en el lisosoma, afectando la función de este organelo, lo cual progresivamente permite manifestaciones renales y extrarrenales (1). En el riñón hay alteración de la función celular, especialmente en el túbulo proximal, lo cual produce poliuria, acidosis metabólica hiperclorémica, glucosuria, fosfaturia, proteinuria y alteración en la síntesis de vitamina D. El compromiso extrarrenal se caracteriza por el depósito corneal de cristales de cistina, observado después de los 12 meses de edad mediante examen oftalmológico con lámpara de hendidura. La acumulación de esta sustancia en las células foliculares tiroideas lleva a fibrosis y atrofia con hipotiroidismo. Otras alteraciones endocrinológicas descritas son la insuficiencia pancreática y el hipogonadismo. El deterioro neurológico es progresivo (7).
Se propone que la historia natural de la enfermedad sin tratamiento conduce en el primer año a tubulopatía proximal, depósitos de cistina en córnea y retraso en el neurodesarrollo. También hay fotofobia progresiva y la manifestación endocrinológica inicial es el hipotiroidismo hacia los 4 o 5 años de vida. La necesidad de terapia de reemplazo renal y/o trasplante renal se hace necesaria entre los 7 y 10 años. A partir de la segunda década de la vida, las manifestaciones oculares se acentúan, lo que acarrea ceguera. Además, aparecen las manifestaciones endocrinológicas, como diabetes e hipogonadismo. El compromiso del sistema nervioso y la miopatía son progresivos (8, 9).
El tratamiento de estos pacientes requiere de un equipo multidisciplinario para la atención de las manifestaciones sistémicas de esta enfermedad. El uso de cisteamina se propone para prevenir el depósito de nuevos cristales de cistina, sin afectar los acúmulos previos, lo cual hace la progresión de la enfermedad más lenta. Este medicamento se conjuga con la cistina para formar un complejo cisteamina – cisteína que sale del lisosoma por medio de un exportador catiónico de aminoácidos PQLC2 (1). Para el tratamiento de los cristales en córnea deben aplicarse gotas de cisteamina. Con el objetivo de disminuir la poliuria, algunos autores sugieren el uso de indometacina en dosis bajas. De manera similar, se ha propuesto administrar carnitina para tratamiento de la miopatía (9). En la actualidad no hay tratamiento definitivo para esta enfermedad, aunque se adelantan investigaciones en modelos
murinos con trasplante de células madre hematopoyéticas con el gen CTNS funcional (10).
La mutación en CTNS: c.926G>T p.G309V (NM_004937) encontrada en la paciente, fue identificada como una variante patogénica por Macías-Vidal y colaboradores en un paciente de 11 años con SF (11), lo que implica cambio de un aminoácido por otro y una proteína con función predicha anormal y ocurrencia de cistinosis. No se realizó estudio a los progenitores.
Respecto a la literatura nacional, en 2015 Soler describe 2 casos de cistinosis nefropática infantil desde el punto de vista de la oftalmología. El primero, un niño diagnosticado con SF a los 8 meses de edad, que a los 13 meses presenta enfermedad renal crónica estadio 3 y se identifican cristales de cistina en córnea. El estudio genético reveló que era un portador doble heterocigoto de las mutaciones c.18_21delGACT y c.1015G>A en el gen CTNS. El segundo caso corresponde al de una niña diagnosticada a los 23 meses, cuyo análisis genético arrojó que tenía una duplicación patogénica homocigótica en p. Thr216Asn fsx226 en el gen CTNS (12). Para los autores sería muy interesante conocer si otros especialistas colombianos tienen reportes o series de caso sobre esta patología.
Agradecimientos
Se hace explícita la gratitud de los autores con la paciente y sus padres por permitir la documentación de este caso clínico; a la Fundación Clínica Infantil Club Noel por promover la investigación en el centro hospitalario y a la Dra. María Alejandra Palma por sus contribuciones en el campo de la genética.
Conflictos de interés
Los autores certifican que no tienen conflictos de interés. Que los recursos para esta investigación salieron de los propios autores y de la Universidad Libre. No se ha recibido financiamiento por parte de entes externos.
Referencias
1. Cherqui S, Courtoy PJ. The renal Fanconi syndrome in cystinosis: pathogenic insights and therapeutic perspectives. Nat Rev Nephrol. 2017; 13 (2): 115–31.
2. Cistinosis. Disponible en: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=ES&Expert=213. Ultimo acceso 31/01/2017
3. Ariceta G, Aguirre M. Tubulopatías en la infancia, que progresan hacia la enfermedad renal crónica. NefroPlus 2011; 4 (1): 11-8.
4. Nesterova G, Gahl WA. Cystinosis: the evolution of a treatable disease. Pediatr Nephrol. 2013; 28 (1): 51–9.
5. Emma F, Nesterova G, Langman C, Labbé A, Cherqui S, Goodyer P, et al. Nephropathic cystinosis: an international consensus document. Nephrol Dial Transplant 2014; 29 (4): iv87-iv94.
6. Ivanova E, De Leo MG, De Matteis MA, Levtchenko E. Cystinosis: clinical presentation, pathogenesis and treatment. Pediatr Endocrinol Rev. 2014; 12 (1): 176–84.
7. Elmonem MA, Veys KR, Soliman NA, van Dyck M, van den Heuvel LP, Levtchenko E. Cystinosis: a review. Orphanet J Rare Dis. 2016; 11(1): 47.
8. Vester U, Schubeter M, Offner G, Brodehl J. Distal myopathy in nephropathic cystinosis. Pediatr Nephrol. 2000; 14: 36–8.
9. Ariceta G, Antonio J, Fernández-Obispo M, FernándezPolo A, Gámez J, García-Villoria J, et al. Cistinosis en pacientes adolescentes y adultos: recomendaciones para la atención integral de la cistinosis. Nefrología. 2015; 35(3): 304–21.
10. Bäumner S, Weber LT. Nephropathic Cystinosis: Symptoms, Treatment, and Perspectives of a Systemic Disease. Front Pediatr [Internet]. Frontiers Media S.A. 2018; 6: 58.
11. Macías-Vidal J, Rodés M, Hernández-Pérez JM, Vilaseca MA, Coll MJ. Analysis of the CTNS gene in 32 cystinosis patients from Spain. Clin Genet. 2009; 76(5): 486–9
12. Bishop R, Hohennfellner K, Liang H, Leal I, Iarossi G, Biswas S, Ferreira C, Soler E. Ophthalmology cystinosis fórum afternoon sesión. EMJ Nephrol. 2017; 5(20): 10-16
Recibido: 24 de junio de 2018
Aceptado: 27 de julio de 2018
Correspondencia: Angie Milena Cárdenas Silva angie-cardenass@unilibre.edu.co